Getting Started with PyBioMed¶
This document is intended to provide an overview of how one can use the PyBioMed functionality from Python. If you find mistakes, or have suggestions for improvements, please either fix them yourselves in the source document (the .py file) or send them to the mailing list: oriental-cds@163.com and gadsby@163.com.
Installing the PyBioMed package¶
PyBioMed has been successfully tested on Linux and Windows systems. The user could download the PyBioMed package via: https://raw.githubusercontent.com/gadsbyfly/PyBioMed/master/PyBioMed/download/PyBioMed-1.0.zip. The installation process of PyBioMed is very easy:
Note
You first need to install RDKit and pybel successfully.
On Windows:
(1): download the PyBioMed-1.0.zip
(2): extract the PyBioMed-1.0.zip file
(3): open cmd.exe and change dictionary to PyBioMed-1.0 (write the command “cd PyBioMed-1.0” in cmd shell)
(4): write the command “python setup.py install” in cmd shell
On Linux:
(1): download the PyBioMed package (.zip)
(2): extract PyBioMed-1.0.zip
(3): open shell and change dictionary to PyBioMed-1.0 (write the command “cd PyBioMed-1.0” in shell)
(4): write the command “python setup.py install” in shell
Getting molecules¶
The PyGetMol provide different formats to get molecular structures, protein sequence and DNA sequence.
Getting molecular structure¶
In order to be convenient to users, the Getmol module provides the tool to get molecular structures by the molecular ID from website including NCBI, EBI, CAS, Kegg and Drugbank.
>>> from PyBioMed.PyGetMol import Getmol
>>> DrugBankID = 'DB01014'
>>> smi = Getmol.GetMolFromDrugbank(DrugBankID)
>>> print smi
N[C@@H](CO)C(=O)O
>>> smi=Getmol.GetMolFromCAS(casid="50-12-4")
>>> print smi
CCC1(c2ccccc2)C(=O)N(C)C(=N1)O
>>> smi=Getmol.GetMolFromNCBI(cid="2244")
>>> print smi
CC(=O)Oc1ccccc1C(=O)O
>>> smi=Getmol.GetMolFromKegg(kid="D02176")
>>> print smi
C[N+](C)(C)C[C@H](O)CC(=O)[O-]
Reading molecules¶
The Getmol module also provides the tool to read molecules in different formats including SDF, Mol, InChi and Smiles.
Users can read a molecule from string.
>>> from PyBioMed.PyGetMol.Getmol import ReadMolFromSmile
>>> mol = ReadMolFromSmile('N[C@@H](CO)C(=O)O')
>>> print mol
<rdkit.Chem.rdchem.Mol object at 0x0D8D3688>
Users can also read a molecule from a file.
>>> from PyBioMed.PyGetMol.Getmol import ReadMolFromSDF
>>> mol = ReadMolFromSDF('./PyBioMed/test/test_data/test.sdf') #You should change the path to your own real path
>>> print mol
<rdkit.Chem.rdmolfiles.SDMolSupplier at 0xd8d03f0>
Getting protein sequence¶
The GetProtein module provides the tool to get protein sequence by the pdb ID and uniprot ID from website.
>>> from PyBioMed.PyGetMol import GetProtein
>>> GetProtein.GetPDB(['1atp'])
>>> seq = GetProtein.GetSeqFromPDB('1atp.pdb')
>>> print seq
GNAAAAKKGSEQESVKEFLAKAKEDFLKKWETPSQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQ
IEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLK
PENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVS
GKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIR
VXINEKCGKEFTEFTTYADFIASGRTGRRNAIHD
>>> seq = GetProtein.GetProteinSequence('O00560')
>>> print seq
MSLYPSLEDLKVDKVIQAQTAFSANPANPAILSEASAPIPHDGNLYPRLYPELSQYMGLSLNEEEIRANVAVVSGAPLQGQLVA
RPSSINYMVAPVTGNDVGIRRAEIKQGIREVILCKDQDGKIGLRLKSIDNGIFVQLVQANSPASLVGLRFGDQVLQINGENCAG
WSSDKAHKVLKQAFGEKITMTIRDRPFERTITMHKDSTGHVGFIFKNGKITSIVKDSSAARNGLLTEHNICEINGQNVIGLKDS
QIADILSTSGTVVTITIMPAFIFEHIIKRMAPSIMKSLMDHTIPEV
Reading protein sequence¶
>>> from PyBioMed.PyGetMol.GetProtein import ReadFasta
>>> f = open('./PyBioMed/test/test_data/protein.fasta') #You should change the path to your own real path
>>> protein_seq = ReadFasta(f)
>>> print protein
['MLIHQYDHATAQYIASHLADPDPLNDGRWLIPAFATATPLPERPARTWPFFLDGAWVLRPDHRGQRLYRTDTGEAAEIVAAG
IAPEAAGLTPTPRPSDEHRWIDGAWQIDPQIVAQRARDAAMREFDLRMASARQANAGRADAYAAGLLSDAEIAVFKAWAIYQMD
LVRVVSAASFPDDVQWPAEPDEAAVIEQADGKASAGDAAAA',
'MLIHQYDHATAQYIASHLADPDPLNDGRWLIPAFATATPLPERPARTWPFFLDGAWVLRPDHRGQRLYRTDTGEAAEIVAAGI
APEAAGLTPTPRPSDEHRWIDGAWQIDPQIVAQRARDAAMREFDLRMASARQANAGRADAYAAGLLSDAEIAVFKAWAIYQMDL
VRVVSAASFPDDVQWPAEPDEAAVIEQADGKASAGDAAAA']
Getting DNA sequence¶
The GetDNA module provides the tool to get DNA sequence by the Gene ID from website.
>>> from PyBioMed.PyGetMol import GetDNA
>>> seq = GetDNA.GetDNAFromUniGene('AA954964')
>>> print seq
>ENA|AA954964|AA954964.1 op24b10.s1 Soares_NFL_T_GBC_S1 Homo sapiens cDNA clone IMAGE:1577755 3', mRNA sequence.
TTTTAAAATATAAAAGGATAACTTTATTGAATATACAAATTCAAGAGCATTCAATTTTTT
TTTAAGATTATGGCATAAGACAGATCAATGGTAATGGTTTATATATCCTATACTTACCAA
ACAGATTAGGTAGATATACTGACCTATCAATGCTCAAAATAACAAAATGAATACATGTCC
CTAAACTATTTCTGTATTCTATGACTACTAAATGGGAAATCTGTCAGCTGACCACCCACC
AGACTTTTTCCCATAGGAAGTTTGATATGCTGTCATTGATATATACCATTTCTGAATATA
AACCTCTATCTTGGGTCCTTTTCTCTTTGCCTACTTCATTATCTGTCTTCCCAACCCACC
TAAGACTTAGTCAAAACAGGATACAGAGATCTGGATGGCTCTACGCAGAG
Reading DNA sequence¶
>>> from PyBioMed.PyGetMol.GetDNA import ReadFasta
>>> f = open('./PyBioMed/test/test_data/example.fasta') #You should change the path to your own real path
>>> dna_seq = ReadFasta(f)
>>> print dna
['GACTGAACTGCACTTTGGTTTCATATTATTTGCTC']
Pretreating structure¶
The PyPretreat can pretreat the molecular structure, the protein sequence and the DNA sequence.
Pretreating molecules¶
The PyPretreatMol can pretreat the molecular structure. The PyPretreatMol proivdes the following functions:
- Normalization of functional groups to a consistent format.
- Recombination of separated charges.
- Breaking of bonds to metal atoms.
- Competitive reionization to ensure strongest acids ionize first in partially ionize molecules.
- Tautomer enumeration and canonicalization.
- Neutralization of charges.
- Standardization or removal of stereochemistry information.
- Filtering of salt and solvent fragments.
- Generation of fragment, isotope, charge, tautomer or stereochemistry insensitive parent structures.
- Validations to identify molecules with unusual and potentially troublesome characteristics.
The user can diconnect metal ion.
>>> from PyBioMed.PyPretreat.PyPretreatMol import StandardizeMol
>>> from rdkit import Chem
>>> mol = Chem.MolFromSmiles('[Na]OC(=O)c1ccc(C[S+2]([O-])([O-]))cc1')
>>> sdm = StandardizeMol()
>>> mol = sdm.disconnect_metals(mol)
>>> print Chem.MolToSmiles(mol, isomericSmiles=True)
O=C([O-])c1ccc(C[S+2]([O-])[O-])cc1.[Na+]
Pretreat the molecular structure using all functions.
>>> from PyBioMed.PyPretreat import PyPretreatMol
>>> stdsmi = PyPretreatMol.StandardSmi('[Na]OC(=O)c1ccc(C[S+2]([O-])([O-]))cc1')
>>> print stdsmi
O=C([O-])c1ccc(C[S](=O)=O)cc1
Pretreating protein sequence¶
The user can check the protein sequence using the PyPretreatPro. If the sequence is right, the result is the number of amino acids. If the sequence is wrong, the result is 0.
>>> from PyBioMed.PyPretreat import PyPretreatPro
>>> protein="ADGCGVGEGTGQGPMCNCMCMKWVYADEDAADLESDSFADEDASLESDSFPWSNQRVFCSFADEDASU"
>>> print PyPretreatPro.ProteinCheck(protein)
0
>>> from PyBioMed.PyPretreat import PyPretreatPro
>>> protein="ADGCRN"
>>> print PyPretreatPro.ProteinCheck(protein)
6
Pretreating DNA sequence¶
The user can check the DNA sequence using the PyPretreatDNA. If the sequence is right, the result is True. If the sequence is wrong, the result is the wrong word.
>>> from PyBioMed.PyPretreat import PyPretreatDNA
>>> DNA="ATTTAC"
>>> print PyPretreatDNA.DNAChecks(DNA)
True
>>> DNA= "ATCGUA"
>>> print PyPretreatDNA.DNAChecks(DNA)
U
Calculating molecular descriptors¶
The PyBioMed package could calculate a large number of molecular descriptors. These descriptors capture and magnify distinct aspects of chemical structures. Generally speaking, all descriptors could be divided into two classes: descriptors and fingerprints. Descriptors only used the property of molecular topology, including constitutional descriptors, topological descriptors, connectivity indices, E-state indices, Basak information indices, Burden descriptors, autocorrelation descriptors, charge descriptors, molecular properties, kappa shape indices, MOE-type descriptors. Molecular fingerprints contain FP2, FP3, FP4,topological fingerprints, Estate, atompairs, torsions, morgan and MACCS.

The descriptors could be calculated through PyBioMed package
Calculating descriptors¶
We could import the corresponding module to calculate the molecular descriptors as need. There is 14 modules to compute descriptors. Moreover, a easier way to compute these descriptors is construct a PyMolecule object, which encapsulates all methods for the calculation of descriptors.
Calculating molecular descriptors via functions¶
The GetConnectivity() function in the connectivity module can calculate the connectivity descriptors. The result is given in the form of dictionary.
>>> from PyBioMed.PyMolecule import connectivity
>>> from rdkit import Chem
>>> smi = 'CCC1(c2ccccc2)C(=O)N(C)C(=N1)O'
>>> mol = Chem.MolFromSmiles(smi)
>>> molecular_descriptor = connectivity.GetConnectivity(mol)
>>> print molecular_descriptor
{'Chi3ch': 0.0, 'knotp': 2.708, 'dchi3': 3.359, 'dchi2': 2.895, 'dchi1': 2.374, 'dchi0': 2.415, 'Chi5ch': 0.068, 'Chiv4': 1.998, 'Chiv7': 0.259, 'Chiv6': 0.591, 'Chiv1': 5.241, 'Chiv0': 9.344, 'Chiv3': 3.012, 'Chiv2': 3.856, 'Chi4c': 0.083, 'dchi4': 2.96, 'Chiv4pc': 1.472, 'Chiv3c': 0.588, 'Chiv8': 0.118, 'Chi3c': 1.264, 'Chi8': 0.636, 'Chi9': 0.322, 'Chi2': 6.751, 'Chi3': 6.372, 'Chi0': 11.759, 'Chi1': 7.615, 'Chi6': 2.118, 'Chi7': 1.122, 'Chi4': 4.959, 'Chi5': 3.649, 'Chiv5': 1.244, 'Chiv4c': 0.04, 'Chiv9': 0.046, 'Chi4pc': 3.971, 'knotpv': 0.884, 'Chiv5ch': 0.025, 'Chiv3ch': 0.0, 'Chiv10': 0.015, 'Chiv6ch': 0.032, 'Chi10': 0.135, 'Chi4ch': 0.0, 'Chiv4ch': 0.0, 'mChi1': 0.448, 'Chi6ch': 0.102}
The function GetTopology() in the topology module can calculate all topological descriptors.
>>> from PyBioMed.PyMolecule import topology
>>> from rdkit import Chem
>>> smi = 'CCC1(c2ccccc2)C(=O)N(C)C(=N1)O'
>>> mol = Chem.MolFromSmiles(smi)
>>> molecular_descriptor = topology.GetTopology(mol)
>>> print len(molecular_descriptor)
25
The function CATS2D() in the cats2d module can calculate all CATS2D descriptors.
>>> from PyBioMed.PyMolecule.cats2d import CATS2D
>>> smi = 'CC(N)C(=O)[O-]'
>>> mol = Chem.MolFromSmiles(smi)
>>> cats = CATS2D(mol,PathLength = 10,scale = 3)
>>> print cats
{'CATS_AP4': 0.0, 'CATS_AP3': 1.0, 'CATS_AP6': 0.0, 'CATS_AA8': 0.0, 'CATS_AA9': 0.0, 'CATS_AP1': 0.0, ......, 'CATS_AP5': 0.0}
>>> print len(cats)
150
Calculating molecular descriptors via PyMolecule object¶
The PyMolecule class can read molecules in different format including MOL, SMI, InChi and CAS. For example, the user can read a molecule in the format of SMI and calculate the E-state descriptors (316).
>>> from PyBioMed import Pymolecule
>>> smi = 'CCC1(c2ccccc2)C(=O)N(C)C(=N1)O'
>>> mol = Pymolecule.PyMolecule()
>>> mol.ReadMolFromSmile(smi)
>>> molecular_descriptor = mol.GetEstate()
>>> print len(molecular_descriptor)
237
The object can also read molecules in the format of MOL file and calculate charge descriptors (25).
>>> from PyBioMed import Pymolecule
>>> mol = Pymolecule.PyMolecule()
>>> mol.ReadMolFromMol('test/test_data/test.mol') #change path to the real path in your own computer
>>> molecular_descriptor = mol.GetCharge()
>>> print molecular_descriptor
{'QNmin': 0, 'QOss': 0.534, 'Mpc': 0.122, 'QHss': 0.108, 'SPP': 0.817, 'LDI': 0.322, 'QCmin': -0.061, 'Mac': 0.151, 'Qass': 0.893, 'QNss': 0, 'QCmax': 0.339, 'QOmax': -0.246, 'Tpc': 1.584, 'Qmax': 0.339, 'QOmin': -0.478, 'Tnc': -1.584, 'QHmin': 0.035, 'QCss': 0.252, 'QHmax': 0.297, 'QNmax': 0, 'Rnc': 0.302, 'Rpc': 0.214, 'Qmin': -0.478, 'Tac': 3.167, 'Mnc': -0.198}
In order to be convenient to users, the object also provides the tool to get molecular structures by the molecular ID from website including NCBI, EBI, CAS, Kegg and Drugbank.
>>> from PyBioMed import Pymolecule
>>> DrugBankID = 'DB01014'
>>> mol = Pymolecule.PyMolecule()
>>> smi = mol.GetMolFromDrugbank(DrugBankID)
>>> mol.ReadMolFromSmile(smi)
>>> molecular_descriptor = mol.GetKappa()
>>> print molecular_descriptor
{'phi': 5.989303307692309, 'kappa1': 22.291, 'kappa3': 7.51, 'kappa2': 11.111, 'kappam1': 18.587, 'kappam3': 5.395, 'kappam2': 8.378}
The code below can calculate all molecular descriptors except fingerprints.
>>> from PyBioMed import Pymolecule
>>> smi = 'CCOC=N'
>>> mol = Pymolecule.PyMolecule()
>>> mol.ReadMolFromSmile(smi)
>>> alldes = mol.GetAllDescriptor()
>>> print len(alldes)
765
Calculating molecular fingerprints¶
In the fingerprint module, there are eighteen types of molecular fingerprints which are defined by abstracting and magnifying different aspects of molecular topology.
Calculating fingerprint via functions¶
The CalculateFP2Fingerprint() function calculates the FP2 fingerprint.
>>> from PyBioMed.PyMolecule.fingerprint import CalculateFP2Fingerprint
>>> import pybel
>>> smi = 'CCC1(c2ccccc2)C(=O)N(C)C(=N1)O'
>>> mol = pybel.readstring("smi", smi)
>>> mol_fingerprint = CalculateFP2Fingerprint(mol)
>>> print len(mol_fingerprint[1])
103
The CalculateEstateFingerprint() function calculates the Estate fingerprint.
>>> from PyBioMed.PyMolecule.fingerprint import CalculateEstateFingerprint
>>> smi = 'CCC1(c2ccccc2)C(=O)N(C)C(=N1)O'
>>> mol = Chem.MolFromSmiles(smi)
>>> mol_fingerprint = CalculateEstateFingerprint(mol)
>>> print len(mol_fingerprint[2])
79
The function GhoseCrippenFingerprint() in the ghosecrippen module can calculate all ghosecrippen descriptors.
>>> from PyBioMed.PyMolecule.ghosecrippen import GhoseCrippenFingerprint
>>> smi = 'CC(N)C(=O)O'
>>> mol = Chem.MolFromSmiles(smi)
>>> ghoseFP = GhoseCrippenFingerprint(mol)
>>> print ghoseFP
{'S3': 0, 'S2': 0, 'S1': 0, 'S4': 0, ......, 'N9': 0, 'Hal2': 0}
>>> print len(ghoseFP)
110
Calculating fingerprint via object¶
The PyMolecule class can calculate eleven kinds of fingerprints. For example, the user can read a molecule in the format of SMI and calculate the ECFP4 fingerprint (1024).
>>> from PyBioMed import Pymolecule
>>> smi = 'CCOC=N'
>>> mol = Pymolecule.PyMolecule()
>>> mol.ReadMolFromSmile(smi)
>>> res = mol.GetFingerprint(FPName='ECFP4')
>>> print res
(4294967295L, {3994088662L: 1, 2246728737L: 1, 3542456614L: 1, 2072128742: 1, 2222711142L: 1, 2669064385L: 1, 3540009223L: 1, 849275503: 1, 2245384272L: 1, 2246703798L: 1, 864674487: 1, 4212523324L: 1, 3340482365L: 1}, <rdkit.DataStructs.cDataStructs.UIntSparseIntVect object at 0x0CA010D8>)
Calculating protein descriptors¶
PyProtein is a tool used for protein feature calculation. PyProtein calculates structural and physicochemical features of proteins and peptides from amino acid sequence. These sequence-derived structural and physicochemical features have been widely used in the development of machine learning models for predicting protein structural and functional classes, post-translational modification, subcellular locations and peptides of specific properties. There are two ways to calculate protein descriptors in the PyProtein module. One is to directly use the corresponding methods, the other one is firstly to construct a PyProtein class and then run their methods to obtain the protein descriptors. It should be noted that the output is a dictionary form, whose keys and values represent the descriptor name and the descriptor value, respectively. The user could clearly understand the meaning of each descriptor.
Calculating protein descriptors via functions¶
The user can input the protein sequence and calculate the protein descriptors using function.
>>> from PyBioMed.PyProtein import AAComposition
>>> protein="ADGCGVGEGTGQGPMCNCMCMKWVYADEDAADLESDSFADEDASLESDSFPWSNQRVFCSFADEDAS"
>>> AAC=AAComposition.CalculateAAComposition(protein)
>>> print AAC
{'A': 11.94, 'C': 7.463, 'E': 8.955, 'D': 14.925, 'G': 8.955, 'F': 5.97, 'I': 0.0, 'H': 0.0, 'K': 1.493, 'M': 4.478, 'L': 2.985, 'N': 2.985, 'Q': 2.985, 'P': 2.985, 'S': 11.94, 'R': 1.493, 'T': 1.493, 'W': 2.985, 'V': 4.478, 'Y': 1.493}
PyBioMed also provides getpdb to get sequence from PDB website to calculate protein descriptors.
>>> from PyBioMed.PyGetMol import GetProtein
>>> GetProtein.GetPDB(['1atp','1efz','1f88'])
>>> seq = GetProtein.GetSeqFromPDB('1atp.pdb')
>>> print seq
GNAAAAKKGSEQESVKEFLAKAKEDFLKKWETPSQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQ
IEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLK
PENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVS
GKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIR
VXINEKCGKEFTEFTTYADFIASGRTGRRNAIHD
>>> from PyBioMed.PyProtein import CTD
>>> protein_descriptor = CTD.CalculateC(protein)
>>> print protein_descriptor
{'_NormalizedVDWVC2': 0.224, '_PolarizabilityC2': 0.328, '_PolarizabilityC3': 0.179, '_ChargeC1': 0.03, '_PolarizabilityC1': 0.493, '_SecondaryStrC2': 0.239, '_SecondaryStrC3': 0.418, '_NormalizedVDWVC3': 0.179, '_SecondaryStrC1': 0.343, '_SolventAccessibilityC1': 0.448, '_SolventAccessibilityC2': 0.328, '_SolventAccessibilityC3': 0.224, '_NormalizedVDWVC1': 0.522, '_HydrophobicityC3': 0.284, '_HydrophobicityC1': 0.328, '_ChargeC3': 0.239, '_PolarityC2': 0.179, '_PolarityC1': 0.299, '_HydrophobicityC2': 0.388, '_PolarityC3': 0.03, '_ChargeC2': 0.731}
Calculate protein descriptors via object¶
The PyProtein can calculate all kinds of protein descriptors in the PyBioMed.
For example, the PyProtein can calculate DPC.
>>> from PyBioMed import Pyprotein
>>> protein="ADGCGVGEGTGQGPMCNCMCMKWVYADEDAADLESDSFADEDASLESDSFPWSNQRVFCSFADEDAS"
>>> protein_class = Pyprotein.PyProtein(protein)
>>> print len(protein_class.GetDPComp())
400
The PyProtein also provide the tool to get sequence from Uniprot through the Uniprot ID.
>>> from PyBioMed import Pyprotein
>>> from PyBioMed.PyProtein.GetProteinFromUniprot import GetProteinSequence
>>> uniprotID = 'P48039'
>>> protein_sequence = GetProteinSequence(uniprotID)
>>> print protein_sequence
MEDINFASLAPRHGSRPFMGTWNEIGTSQLNGGAFSWSSLWSGIKNFGSSIKSFGNKAWNSNTGQMLRDKLKDQNFQQKVVDGL
ASGINGVVDIANQALQNQINQRLENSRQPPVALQQRPPPKVEEVEVEEKLPPLEVAPPLPSKGEKRPRPDLEETLVVESREPPS
YEQALKEGASPYPMTKPIGSMARPVYGKESKPVTLELPPPVPTVPPMPAPTLGTAVSRPTAPTVAVATPARRPRGANWQSTLNS
IVGLGVKSLKRRRCY
>>> protein_class = Pyprotein.PyProtein(protein_sequence)
>>> CTD = protein_class.GetCTD()
>>> print len(CTD)
147
The PyProtein can calculate all protein descriptors except the tri-peptide composition descriptors.
>>> from PyBioMed import Pyprotein
>>> protein="ADGCGVGEGTGQGPMCNCMCMKWVYADEDAADLESDSFADEDASLESDSFPWSNQRVFCSFADEDAS"
>>> protein_class = Pyprotein.PyProtein(protein)
>>> print len(protein_class.GetALL())
10049
Calculating DNA descriptors¶
The PyDNA module can generate various feature vectors for DNA sequences, this module could:
- Calculating three nucleic acid composition features describing the local sequence information by means of kmers (subsequences of DNA sequences);
- Calculating six autocorrelation features describing the level of correlation between two oligonucleotides along a DNA sequence in terms of their specific physicochemical properties;
- Calculating six pseudo nucleotide composition features, which can be used to represent a DNA sequence with a discrete model or vector yet still keep considerable sequence order information, particularly the global or long-range sequence order information, via the physicochemical properties of its constituent oligonucleotides.
Calculating DNA descriptors via functions¶
The user can input a DNA sequence and calculate the DNA descriptors using functions.
>>> from PyBioMed.PyDNA.PyDNAac import GetDAC
>>> dac = GetDAC('GACTGAACTGCACTTTGGTTTCATATTATTTGCTC', phyche_index=['Twist','Tilt'])
>>> print(dac)
{'DAC_4': -0.004, 'DAC_1': -0.175, 'DAC_2': -0.185, 'DAC_3': -0.173}
The user can check the parameters and calculate the descriptors.
>>> from PyBioMed.PyDNA import PyDNApsenac
>>> from PyBioMed.PyDNA.PyDNApsenac import GetPseDNC
>>> dnaseq = 'GACTGAACTGCACTTTGGTTTCATATTATTTGCTC'
>>> PyDNApsenac.CheckPsenac(lamada = 2, w = 0.05, k = 2)
>>> psednc = GetPseDNC('ACCCCA',lamada=2, w=0.05)
>>> print(psednc)
{'PseDNC_18': 0.0521, 'PseDNC_16': 0.0, 'PseDNC_17': 0.0391, 'PseDNC_14': 0.0, 'PseDNC_15': 0.0, 'PseDNC_12': 0.0, 'PseDNC_13': 0.0, 'PseDNC_10': 0.0, 'PseDNC_11': 0.0, 'PseDNC_4': 0.0, 'PseDNC_5': 0.182, 'PseDNC_6': 0.545, 'PseDNC_7': 0.0, 'PseDNC_1': 0.0, 'PseDNC_2': 0.182, 'PseDNC_3': 0.0, 'PseDNC_8': 0.0, 'PseDNC_9': 0.0}
Calculating DNA descriptors via object¶
The PyDNA can calculate all kinds of protein descriptors in the PyBioMed.
For example, the PyDNA can calculate SCPseDNC.
>>> from PyBioMed import Pydna
>>> dna = Pydna.PyDNA('GACTGAACTGCACTTTGGTTTCATATTATTTGCTC')
>>> scpsednc = dna.GetSCPseDNC()
>>> print len(scpsednc)
16
Calculating Interaction descriptors¶
The PyInteraction module can generate six types of interaction descriptors indcluding chemical-chemical interaction features, chemical-protein interaction features, chemical-DNA interaction features, protein-protein interaction features, protein-DNA interaction features, and DNA-DNA interaction features by integrating two groups of features.
The user can choose three different types of methods to calculate interaction descriptors. The function CalculateInteraction1() can calculate two interaction features by combining two features.
The function CalculateInteraction2() can calculate two interaction features by two multiplied features.
The function CalculateInteraction3() can calculate two interaction features by
The function CalculateInteraction3() is only used in the same type of descriptors including chemical-chemical interaction, protein-protein interaction and DNA-DNA interaction.
The user can calculate chemical-chemical features using three methods .
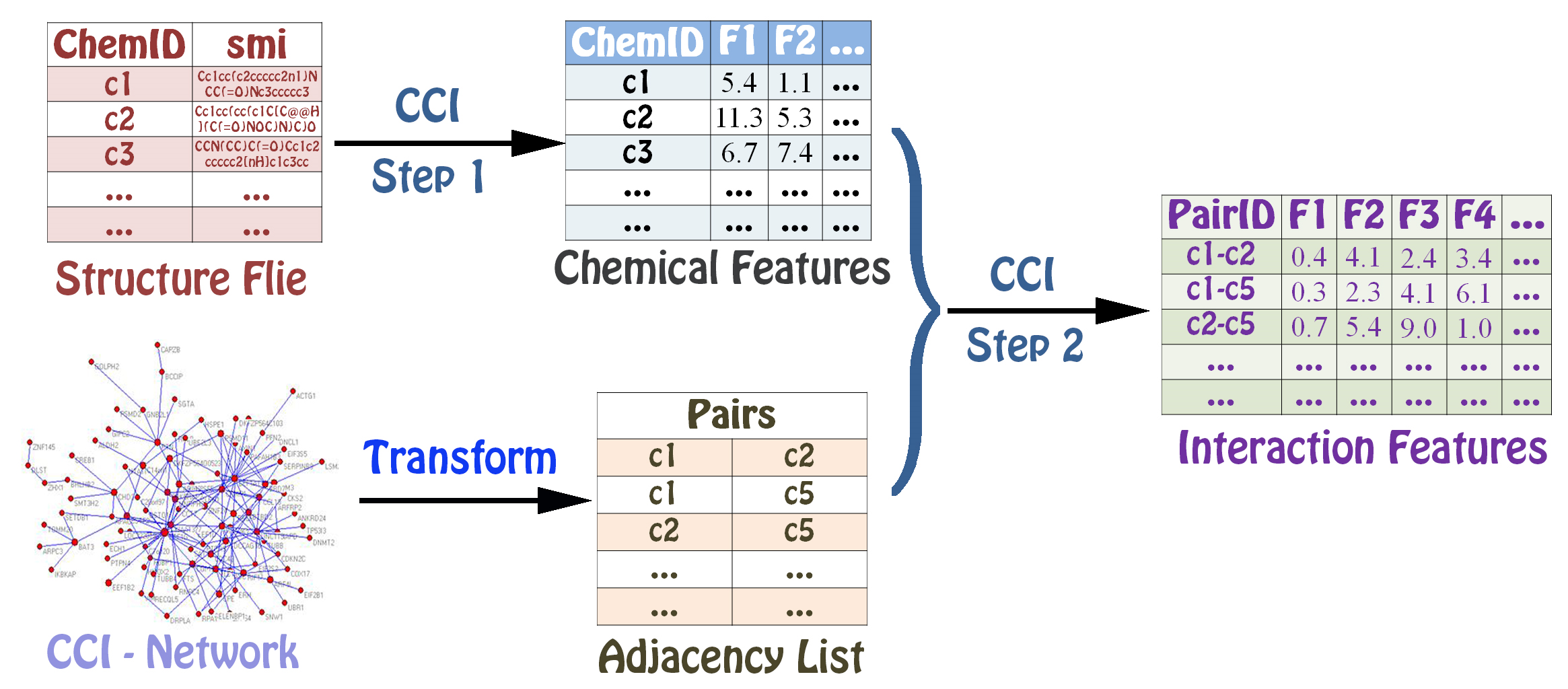
The calculation process for chemical-chemical interaction descriptors.
>>> from PyBioMed.PyInteraction import PyInteraction
>>> from PyBioMed.PyMolecule import moe
>>> from rdkit import Chem
>>> smis = ['CCCC','CCCCC','CCCCCC','CC(N)C(=O)O','CC(N)C(=O)[O-].[Na+]']
>>> m = Chem.MolFromSmiles(smis[3])
>>> mol_des = moe.GetMOE(m)
>>> mol_mol_interaction1 = PyInteraction.CalculateInteraction1(mol_des,mol_des)
>>> print mol_mol_interaction1
{'slogPVSA6ex': 0.0, 'PEOEVSA10ex': 0.0,......, 'EstateVSA9ex': 4.795, 'slogPVSA2': 4.795, 'slogPVSA3': 0.0, 'slogPVSA0': 5.734, 'slogPVSA1': 17.118, 'slogPVSA6': 0.0, 'slogPVSA7': 0.0, 'slogPVSA4': 6.924, 'slogPVSA5': 0.0, 'slogPVSA8': 0.0, 'slogPVSA9': 0.0}
>>> print len(mol_mol_interaction1)
120
>>> mol_mol_interaction2 = PyInteraction.CalculateInteraction2(mol_des,mol_des)
>>> print len(mol_mol_interaction2)
3600
>>> mol_mol_interaction3 = PyInteraction.CalculateInteraction3(mol_des,mol_des)
{'EstateVSA9*EstateVSA9': 22.992, 'EstateVSA9+EstateVSA9': 9.59, 'PEOEVSA1+PEOEVSA1': 9.59, 'VSAEstate10*VSAEstate10': 0.0, 'PEOEVSA3*PEOEVSA3': 0.0, 'PEOEVSA11*PEOEVSA11': 0.0, 'PEOEVSA4*PEOEVSA4': 0.0, 'VSAEstate2+VSAEstate2': 0.0, 'MRVSA0+MRVSA0': 19.802, 'MRVSA6+MRVSA6': 0.0......}
>>> print len(mol_mol_interaction3)
120
The user can calculate chemical-protein feature using two methods.
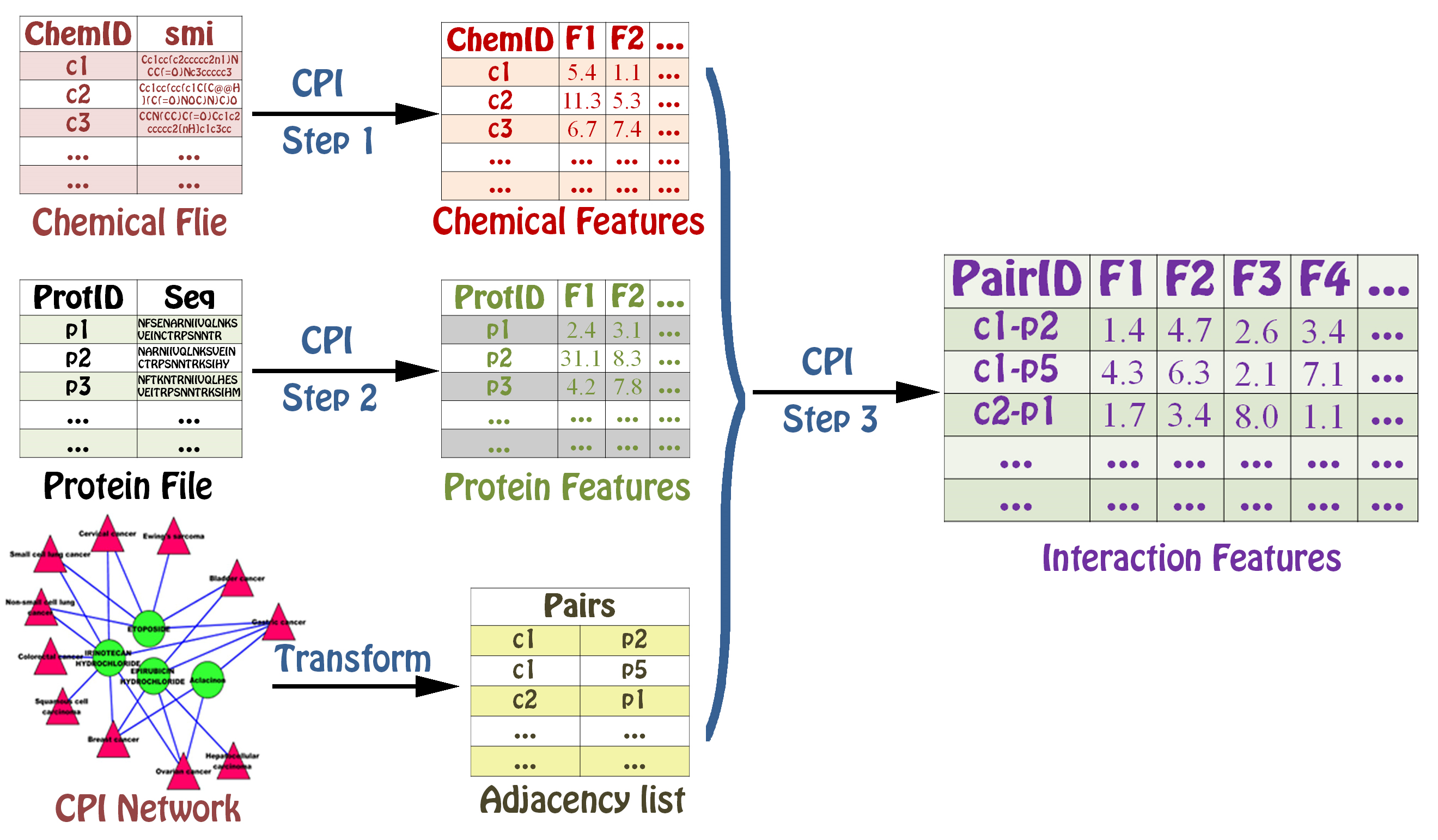
The calculation process for chemical-protein interaction descriptors.
>>> from rdkit import Chem
>>> from PyBioMed.PyMolecule import moe
>>> from PyBioMed.PyInteraction.PyInteraction import CalculateInteraction2
>>> smis = ['CCCC','CCCCC','CCCCCC','CC(N)C(=O)O','CC(N)C(=O)[O-].[Na+]']
>>> m = Chem.MolFromSmiles(smis[3])
>>> mol_des = moe.GetMOE(m)
>>> from PyBioMed.PyDNA.PyDNApsenac import GetPseDNC
>>> protein_des = GetPseDNC('ACCCCA',lamada=2, w=0.05)
>>> pro_mol_interaction1 = PyInteraction.CalculateInteraction1(mol_des,protein_des)
>>> print len(pro_mol_interaction1)
78
>>> pro_mol_interaction2 = CalculateInteraction2(mol_des,protein_des)
>>> print len(pro_mol_interaction2)
1080
The user can calculate chemical-DNA feature using two methods.
>>> from PyBioMed.PyDNA import PyDNAac
>>> DNA_des = PyDNAac.GetTCC('GACTGAACTGCACTTTGGTTTCATATTATTTGCTC', phyche_index=['Dnase I', 'Nucleosome','MW-kg'])
>>> from rdkit import Chem
>>> smis = ['CCCC','CCCCC','CCCCCC','CC(N)C(=O)O','CC(N)C(=O)[O-].[Na+]']
>>> m = Chem.MolFromSmiles(smis[3])
>>> mol_des = moe.GetMOE(m)
>>> mol_DNA_interaction1 = PyInteraction.CalculateInteraction1(mol_des,DNA_des)
>>> print len(mol_DNA_interaction1)
72
>>> mol_DNA_interaction2 = PyInteraction.CalculateInteraction2(mol_des,DNA_des)
>>> print len(mol_DNA_interaction2)
720